
タンパク質立体構造推論ソフトウェア「OpenFold」を富岳に実装、利用可能に 解析スピード高速化も達成

理化学研究所(理研)は、タンパク質立体構造推論ソフトウエア「OpenFold」をスーパーコンピュータ「富岳」へ実装したと発表した。オープンソースソフトウェアとして広く利用できる。また高速化手法の開発により高スループットも達成したという。
独自の改良で処理スピードを改善
今回「富岳」に実装された「OpenFold」は、英国DeepMind社(Googleを核としたAlphabetグループのAI開発企業)により開発され、実験的に決定された約20万個のタンパク質立体構造とそれに対応するアミノ酸配列を学習することで、構造未知のアミノ酸配列から立体構造モデルを推論するソフトウェア「AlphaFold2」をオープンソース化したもの。
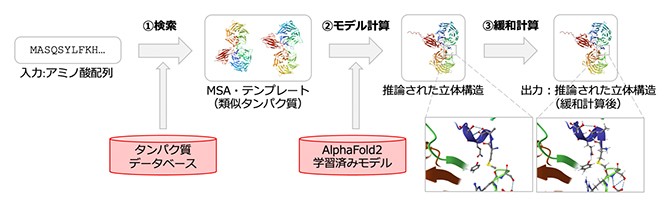
「AlphaFold2」における推論は、アミノ酸配列を入力とした以下の手順の演算からなるという。
①タンパク質データベースを用いたMSA※1、テンプレート検索※2
②「AlphaFold2」内で構築した学習済みモデルを用いたモデル計算※3
③緩和計算※4(物理化学的な構造ゆがみを解消するための分子力場を用いた構造最適化)
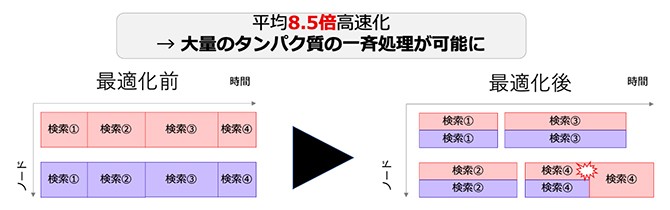
理研の研究チームは今回、「OpenFold」を実装する際、検索タスクの実行順序および同時実行数を最適化し、メモリ不足時には計算資源量を自動で再割り当てする機構を構築することで、構築前に比べて平均8.5倍の高速化(スループット増加)を実現した。
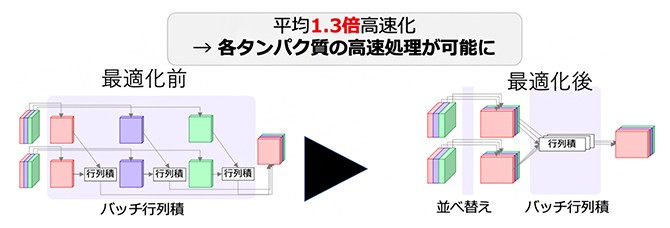
モデル計算タスクにおいては、バッチ行列積※6の入力全体をあらかじめ並べ替え性能低下を回避する工夫を行なったり、アテンション機構※7など一部のGPU向け実装をCPUに移植するなどしてタスクを最適化した結果、最適化前に比べて平均1.3倍の高速化を実現したという。このように高速化を施した「富岳」実装版「OpenFold」を用い、1万件の入力アミノ酸配列を評価したところ、推論手順全体で最適化前と比較し6.3倍の高スループットを達成した。
理研は現在、「富岳」への創薬DXプラットフォーム※7の構築を進めており、今回開発した「富岳」実装版「OpenFold」は、創薬DXプラットフォームにおいて、標的タンパク質の構造を高速に推論する重要な要素技術となるとしている。また並行してクラウド上で「富岳」を直接利用できる「バーチャル富岳」の環境整備も進めていることから、今後そちらへの実装も視野に入れて取り組むとしている。
なお「富岳」実装版「OpenFold」は以下の理研のGitHubで公開されており、創薬研究者などが自ら自身の環境へインストール可能となっている。
※1 MSA
複数のアミノ酸配列に対してギャップを挿入して対応する部分が並ぶように整列し、アミノ酸配列の一致度・類似度を調べる方法。類似性が高いアミノ酸配列を持つタンパク質の機能や構造は似ていることが多いことから、未知のタンパク質の機能や構造に対して示唆を与える。MSAはMultiple Sequence Alignmentの略。
※2 テンプレート検索
アミノ酸から立体構造を予測する際に、鋳型となる構造を既知構造のデータベースから検索すること。鋳型構造の使用は任意であり、推論の際に必ずしも必要ではない。
※3 モデル計算
全てのアミノ酸残基が原点に集中している構造を初期構造として、アミノ酸配列の特徴を共進化の観点から表すMSA表現、およびアミノ酸残基対の距離の特徴を表すペア表現を手掛かりに、各アミノ残基に対して相対的移動(並進・回転)を繰り返すことで立体構造を得る。
※4 緩和計算
物理化学的な構造ゆがみを解消するため、推論した立体構造に対して軽微な分子シミュレーションを実施すること。
※5 バッチ行列積
一定量のデータ(バッチ)に対して、まとめて行列の積を計算すること。
※6 アテンション機構
ニューラルネットワークの学習法の一つで、認知的な注意を模倣し、データの重要な部分に注意を払う仕組み。